A multi-GPU GROMACS replica ensemble in your own AWS
We built a GPU GROMACS-with-MPI container (none ships upstream) and ran Trp-cage temperature-REMD across 4 A10G GPUs of one spot node, with replica exchange working at ~53% acceptance. A throughput-and-operating-model demo, not a converged folding study, in your own AWS account.
Published June 3 2026. Numbers are from runs on Clusterra's dev cluster (June 3 2026): GROMACS 2024.4 + CUDA (A10G
sm_86), OpenMPI, on EC2 spot. A dedicated single-GPU throughput benchmark (5 ns,-resethway); a 4-replica REMD run (one sim/GPU); and an 8-replica REMD run (two sims/GPU). This demonstrates the execution pattern, throughput, replica-exchange behaviour, and cloud economics of single-node multi-GPU GROMACS — not a converged folding result. A real Trp-cage T-REMD study needs ~40 replicas and >100 µs (Day et al. 2010, on a near-identical system); ours is many orders of magnitude shorter. Full config and honest caveats are in Honest scope.
A multi-GPU GROMACS replica ensemble in your own AWS
Eight GROMACS GPU simulations exchanging temperatures across the four A10Gs of a single g5.12xlarge on spot, launched as one managed Slurm job inside the customer's own AWS account — using a GROMACS image we had to build ourselves, because none exists upstream.
A computational chemist wants to run an ensemble of GPU MD simulations — replica exchange, an FEP λ-set, or just N independent trajectories — on hardware they don't own or administer. On Clusterra that is one Slurm submission against a managed cluster in their own cloud account: the right container is pulled, the A10G GPUs each run a simulation via GROMACS' -multidir, all on spot, no cluster to stand up and no data egress. This post asks the narrow, honest question: can we run a real multi-GPU MPI GROMACS workload here, how fast and how cheap, does replica exchange actually work, and how does that line up with the literature.
We kept this single-node on purpose. Multi-node GROMACS is the wrong tool on commodity cloud networking: without a low-latency fabric (EFA/InfiniBand), GROMACS' strong scaling collapses past ~2 nodes, and the cost-optimal pattern is instead one independent simulation per GPU, packed onto a single box.
TL;DR
- We had to build the image. No upstream container ships a GPU build of GROMACS with real MPI: the NGC (
nvcr.io/hpc/gromacs) and Docker Hub (gromacs/gromacs) GPU images are thread-MPI only — nogmx_mpibinary, somdrun -multidir(multi-simulation / replica exchange) can't run on them. We builtgmx_mpi+ CUDA from source through our normal image CI and pushed it to our registry; the cluster pulls it like any other template image. - The pattern:
gmx_mpi mdrun -multidirran N replicas across the 4 A10Gs of one g5.12xlarge — single-node multi-GPU MPI, entirely within one pod, no inter-node network. This is the regime the canonical cloud-GROMACS benchmark (Kutzner et al. 2022) found most cost-effective: one sim per GPU beats decomposing a single sim across a box's GPUs by ~4×. - System: Trp-cage (PDB 1L2Y), a 20-residue mini-protein, in explicit TIP3P water — 8,307 atoms (304 protein + 2,662 waters + 17 ions), amber99sb-ildn, 2 fs.
- Single-GPU throughput (clean benchmark): ~1,547 ns/day on one A10G (5 ns,
-resethway, 1 rank / 3 OpenMP threads,-nb gpu -pme gpu). Inside the REMD runs, replicas clocked ~1,365 ns/day (one sim/GPU) and ~536 ns/day (two sims/GPU sharing a card). - Replica exchange works — with a dense enough ladder. A coarse 4-replica / 11 K ladder gave 0% acceptance (expected); a dense 8-replica / ~2.1 K ladder gave ~53% acceptance and every replica visited all 8 temperature rungs. The contrast is the point.
- Cost: g5.12xlarge spot in us-east-1 was $2.38/hr the day of the run (lowest AZ; $2.5–2.9 typical) vs $5.672 on-demand → roughly $9–13 per microsecond of aggregate sampling on spot (extrapolated from steady-state rate; see below).
No upstream image ships this — so we built one
GROMACS has two parallelism layers. Thread-MPI is a single-node, in-process build (gmx) — fine for one simulation on one or more GPUs. Real MPI (gmx_mpi, built against OpenMPI/MPICH) is what mdrun -multidir needs: many simulations at once, one MPI rank per simulation — how GROMACS does replica exchange and multi-simulation ensembles.
Every prebuilt GPU GROMACS image we checked ships thread-MPI only — the NGC HPC image and the Docker Hub gromacs/gromacs tags both lack a gmx_mpi binary. (Same trap as the stock RELION container.) So a -multidir ensemble simply can't launch on them.
So we did the boring, correct thing: a Dockerfile (nvidia/cuda:12.6-devel base → GROMACS 2024.4 with -DGMX_MPI=ON -DGMX_GPU=CUDA, pinned to A10G's sm_86) built by our existing image CI and pushed to our registry. The cluster pulls it by tag like any other template image — build artifact equals run artifact, one surface. (Two gotchas so others don't hit them: GROMACS' bundled-FFTW build is incompatible with the Ninja generator — use Make; and a standard nvidia/cuda base avoids the non-standard CUDA library layout the NVIDIA HPC-SDK image scatters across cuda/, math_libs/, and targets/ dirs.)
The run
One managed Slurm submission asked for --gres=gpu:a10g:4 on the GPU partition; the autoscaler brought up a g5.12xlarge on spot and the job pulled the GROMACS image (cached as a SIF on the shared filesystem). System prep ran as ordinary single-process gmx_mpi calls inside the container — pdb2gmx (amber99sb-ildn / TIP3P) → editconf → solvate → genion (0.15 M NaCl, neutralized) → GPU energy minimization — giving 8,307 atoms. Then the replicas launched together (this is the 8-replica run):
apptainer exec --nv $SIF \
mpirun --mca ras ^slurm --oversubscribe -np 8 \
gmx_mpi mdrun -multidir r0 r1 r2 r3 r4 r5 r6 r7 -deffnm md \
-replex 500 -ntomp 2 -nb gpu -pme gpu
Two practical notes. MPI launch: Slurm hands the batch step a single task, so OpenMPI must be told to ignore Slurm's allocator and oversubscribe (--mca ras ^slurm --oversubscribe) to fork the eight ranks locally within the one pod; because it's single-node, all MPI traffic stays inside the pod. CPU: these runs are GPU-bound, so a few OpenMP threads per rank saturate each A10G — the job deliberately requests only the CPU it needs rather than the whole box. GROMACS auto-mapped the ranks onto the four A10Gs (one per GPU in the 4-replica run; two per GPU in the 8-replica run).
How fast, honestly
A dedicated single-GPU benchmark — 5 ns, -resethway (which resets the timer past the PME-tuning/load-balancing warmup) — gave 1,547 ns/day on one A10G for this 8,307-atom system (Performance: 1546.8 ns/day, 0.016 h/ns, 1 MPI rank, 3 OpenMP threads). Inside the REMD runs the per-replica rate was lower and startup-affected: ~1,365 ns/day at one sim per GPU (a short 0.3 ns run with tuning still in the window), and ~536 ns/day when two replicas share a card.
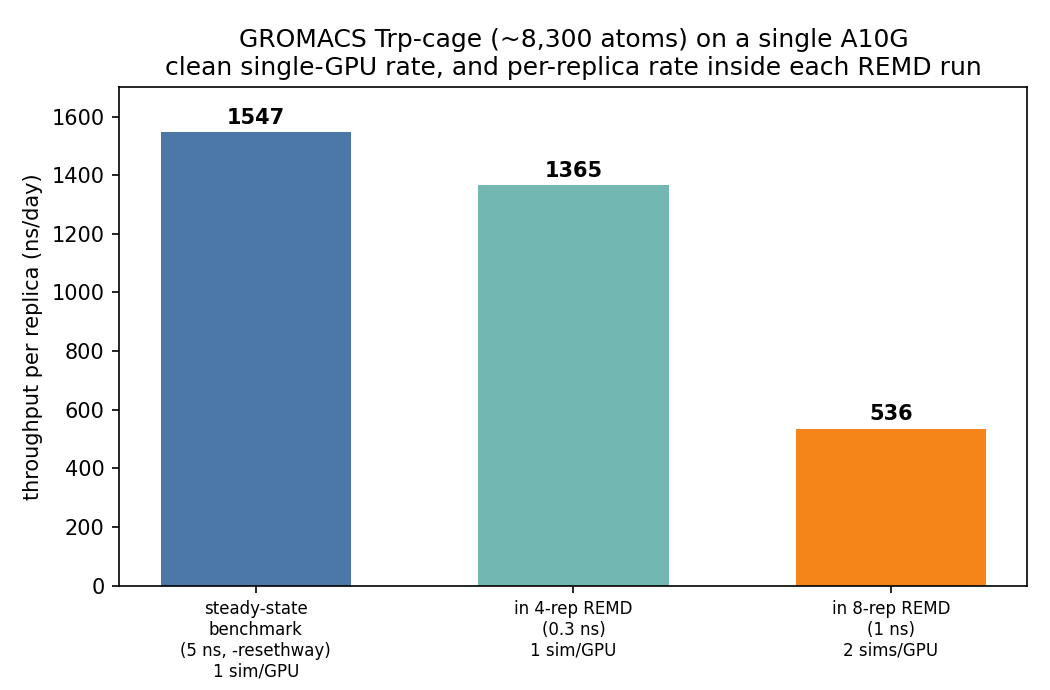
That per-GPU number deserves scrutiny, because there is no published A10G GROMACS figure to cite — the A10G postdates the major benchmark papers. The honest framing is triangulation, with the caveat that published single-GPU numbers use far larger systems: Kutzner et al. 2022 report benchMEM (82,000 atoms, 2 fs) at 58 / 101 / 130 ns/day on T4 / V100 / A100. Our system is ~10× smaller, and GROMACS single-GPU throughput rises with the inverse of atom count down to a per-GPU efficiency floor near ~10,000 atoms — which is what puts an ~8k-atom system into four-figure ns/day. The A10G itself is roughly a V100-class card for MD (somewhat below an A100). So ~1,547 ns/day for a system this small is plausible and consistent — but it is a small-system number, not comparable to the 82k-atom benchmarks, and we'd treat any cross-GPU comparison as indicative only.
The economics
The defensible, differentiated result is the single-node multi-GPU pattern and its cost. Kutzner et al. (2022) — the canonical cloud-GROMACS benchmark (a Max Planck study that runs on AWS) — found that one independent simulation per GPU via -multidir delivers "about four times" the cost-efficiency of decomposing a single simulation across a multi-GPU box, because PME scaling across GPUs needs a fast interconnect; without one, one-sim-per-GPU wins. That is exactly our pattern.
At the day's $2.38/hr g5.12xlarge spot price, four A10Gs at the steady-state rate is ~6,200 ns/day aggregate → ~$9 per microsecond of sampling; the 8-replica REMD (two sims/GPU, ~4,300 ns/day aggregate) is ~$13/µs (roughly $22 and $32/µs at on-demand). These are extrapolated from steady-state ns/day rather than a long measured run, and are a conservative upper bound — at production length the fixed startup/tuning overhead amortizes away. For context, the same paper put cost-optimized FEP at ~$16 per free-energy edge on single-GPU g4dn/g5 instances — the same economical regime, here in the customer's own account, on spot, with no cluster to administer.
Does the replica exchange actually work?
Yes — once the ladder is dense enough, and the two runs make a clean teaching point.
The 4-replica run used 300 / 311 / 322 / 334 K (11 K spacing) and got 0 accepted exchanges out of 149 attempts: at that spacing the replicas' potential-energy distributions barely overlap for an ~8,300-atom explicit-solvent system, so no swaps pass the Metropolis test. It behaves as four independent trajectories with the exchange machinery attached — the textbook outcome for too few replicas, not a malfunction.
The 8-replica run used a dense geometric ladder, 300.0 → 315.0 K (~2.1 K spacing), and exchange worked. GROMACS reported a mean acceptance probability of ~0.53 across all seven neighbour pairs (its "average probabilities" and "average number of exchanges" lines agree at ~0.53), over 999 attempts. The empirical transition matrix below shows off-diagonals of ~0.26–0.28 — about half the acceptance figure, because GROMACS alternates odd/even neighbour pairs each step, so any given pair is offered a swap only every other attempt. Both numbers describe the same healthy exchange.
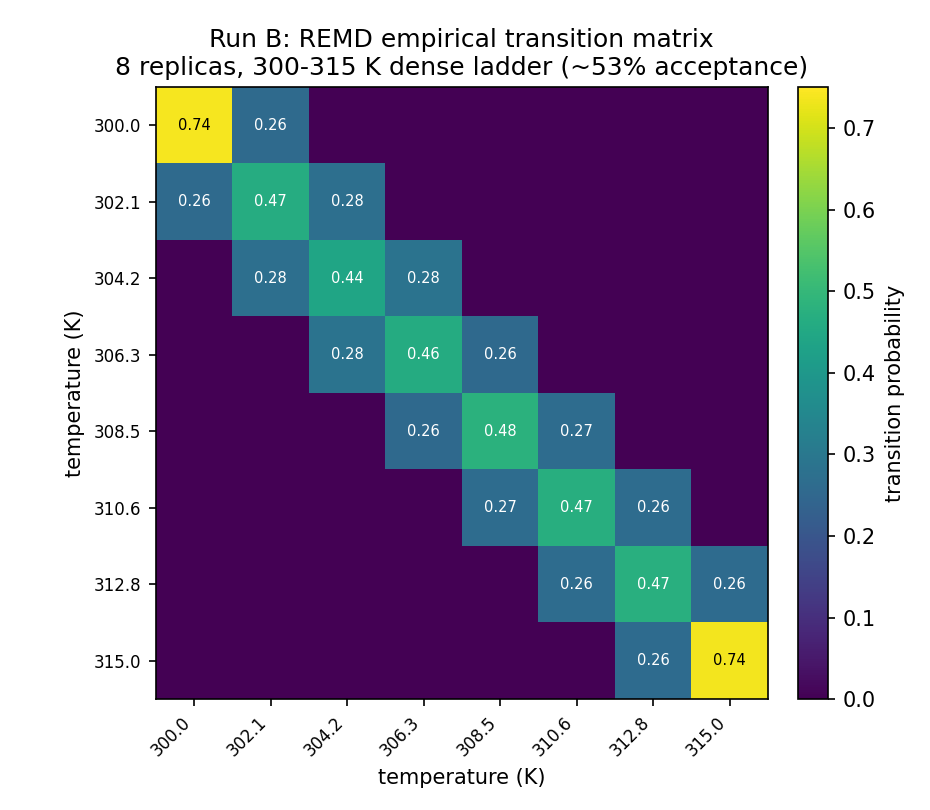
A tridiagonal transition matrix with no gaps means probability can random-walk the full temperature range — and it did: de-multiplexing the exchange record shows every one of the 8 replicas visited all 8 temperature rungs within the 1 ns run.
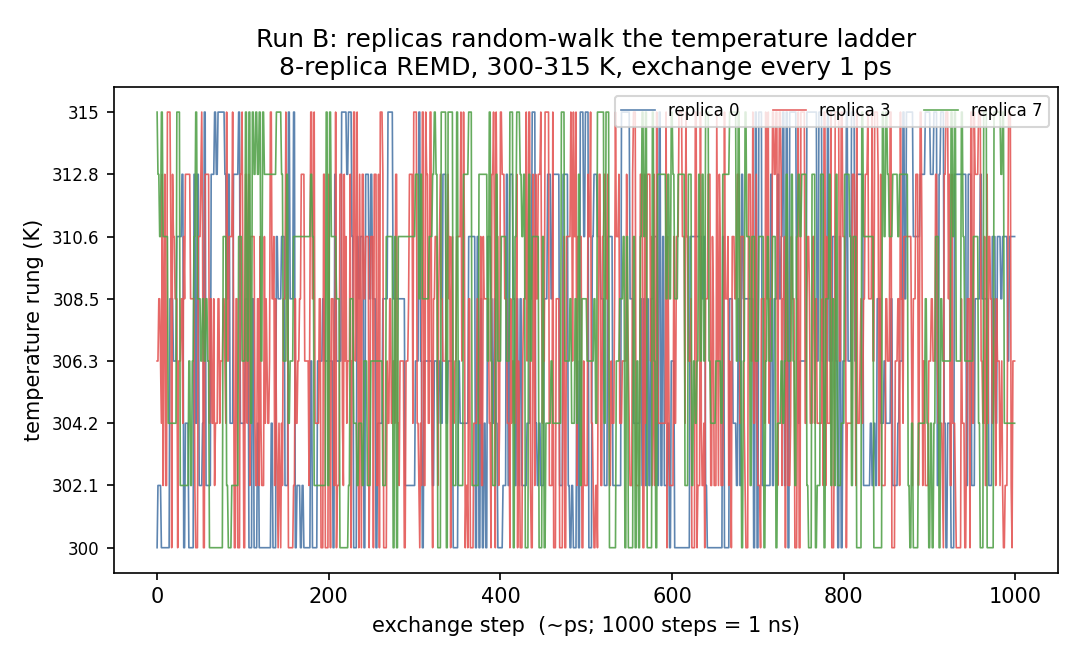
Honest nuance: ~0.53 acceptance is actually above the commonly-cited ~0.2 optimum — our 2.1 K spacing is slightly over-resolved, so a production ladder (e.g. via the Patriksson–van der Spoel predictor) would space a touch wider and need fewer replicas for the same coverage. But the useful result is the dependence itself: 0% at 11 K → ~53% at 2.1 K, demonstrated on real hardware in minutes.
Honest scope
These are short runs that validate an execution pattern, its throughput/economics, and that replica exchange functions. They are not science:
- 8 replicas / 1 ns is not a folding study. The closest published comparator — Day, Paschek & García (2010), Trp-cage in ~2,635 TIP3P waters (a similarly-sized ~8k-atom explicit-solvent system) — used 40 replicas over 280–540 K and >100 µs. We are many orders of magnitude short and fold nothing.
- Our dense ladder is over-resolved, not under (≈0.53 acceptance vs the ~0.2 optimum); a real campaign would tune the ladder and run far longer.
- Throughput is one configuration each (2 fs, h-bond constraints, no HMR, PME on GPU); HMR/4 fs, larger systems, or separate-PME tuning will move the numbers. Costs are extrapolated from rate, not a long measured run.
- No accuracy or convergence claim. Nothing here speaks to force-field accuracy, folding free energies, or sampling convergence.
What it does establish: the architecture runs a genuine multi-GPU MPI GROMACS workload — image built through normal CI, pulled into a managed Slurm job, the box's GPUs each running independent simulations via gmx_mpi -multidir, replica exchange mixing across the full ladder, on spot, in the customer's AWS account — at throughput and cost consistent with the published cloud-GROMACS literature.
Why this matters
The capability isn't a benchmark number; it's that a single scientist runs a multi-GPU GROMACS ensemble without owning or administering a cluster, on spot in their own account, through one Slurm submission — including the part that usually blocks this outright: there was no suitable container, so the platform builds and ships one through the same CI that ships everything else, and the cluster pulls it by tag. The orchestration the scientist didn't have to write — autoscaling the GPU box, pulling and caching the image, mapping ranks to GPUs, recovering spot — is the product. Replica exchange today, an FEP λ-ensemble or a batch of independent production runs tomorrow: same -multidir pattern, same one-per-GPU economics (or two-per-GPU when you need more concurrency), same managed cluster, same account. This launch pattern will ship as a catalog template.
Run it in your own AWS
Multi-GPU GROMACS replica ensembles — and the rest of your HPC stack — run on a managed Slurm cluster in your own AWS account: on spot, no cluster to stand up, no data egress. Start at clusterra.cloud, or email hello@clusterra.cloud.
Appendix — exact configuration
- Image: GROMACS 2024.4, built from
nvidia/cuda:12.6.3-devel-ubuntu22.04with-DGMX_MPI=ON -DGMX_GPU=CUDA -DGMX_CUDA_TARGET_SM=86 -DGMX_BUILD_OWN_FFTW=ON -DGMX_SIMD=AVX2_256, OpenMPI 4.1.2; built and published by the project's image CI. - Hardware: 1× AWS g5.12xlarge = 4× NVIDIA A10G (24 GB), EC2 Spot, us-east-1 (single-GPU benchmark ran on 1× A10G / g5.xlarge).
- System: Trp-cage (PDB 1L2Y, model 1), amber99sb-ildn, TIP3P, cubic box (1.0 nm padding), 0.15 M NaCl neutralized; 8,307 atoms.
- Common protocol: steepest-descent minimization on GPU; production
md, 2 fs,cutoff-scheme=Verlet, PME,rlist=rcoulomb=rvdw=1.0 nm,v-rescalethermostat, no pressure coupling,constraints=h-bonds, per-replica generated velocities. - Throughput benchmark: 1 replica, 1× A10G, 5 ns,
-resethway -ntomp 3 -nb gpu -pme gpu→ 1,546.8 ns/day (0.016 h/ns). - 4-replica REMD: 300 / 311 / 322 / 334 K, one per GPU,
-ntomp 3 -replex 1000, 0.3 ns/replica → ~1,365 ns/day/replica; 149 attempts, 0 accepted. - 8-replica REMD: geometric ladder 300.0–315.0 K, two per GPU,
-ntomp 2 -replex 500, 1 ns/replica → ~536 ns/day/replica, ~4,300 ns/day aggregate; 999 attempts, ~0.53 mean acceptance, tridiagonal transition matrix, all replicas traversing the full ladder. - Launch:
gmx_mpi mdrun -multidirvia the container's OpenMPI,--mca ras ^slurm --oversubscribe; single node, intra-pod MPI only.
References
- Kutzner et al., GROMACS in the Cloud (J. Chem. Inf. Model. 2022), PMC9006219 — single-GPU benchmark numbers, the
-multidircost-efficiency finding, cloud FEP costs (a Max Planck study run on AWS). - NHR@FAU, GROMACS performance on different GPU types (2022) — single-GPU ns/day across GPU models.
- MPI benchmark set (benchMEM 82k atoms): mpinat.mpg.de/grubmueller/bench.
- Day, Paschek & García, Microsecond simulations of the folding/unfolding thermodynamics of the Trp-cage (Proteins 2010), PMC3534748 — 40-replica explicit-solvent T-REMD of the size-matched system.
- Patriksson & van der Spoel, A temperature predictor for parallel tempering simulations (Phys. Chem. Chem. Phys. 2008) — replica-count / acceptance theory.
- AWS EC2 pricing (g5.12xlarge on-demand $5.672/hr); g5.12xlarge us-east-1 spot $2.38/hr (lowest AZ) on the run date.